INTRODUCCION
Se aplica el nombre de grasas o lípidos a una serie de compuestos que
tienen en común el ser solubles en determinados disolventes orgánicos e
insolubles en el agua, por lo que deben de modificarse físicamente para poder
ser absorbidos por la pared del intestino. Esta absorción es tanto más
fácil cuanto menor es el punto de fusión, y más aún si éste es inferior a la
temperatura corporal (37ºC).
Los lípidos constituyen el grupo de biomoléculas menos caracterizables
desde el punto de vista químico, ya que la única propiedad que comparten es su
insolubilidad en agua y su capacidad de disolverse en solventes orgánicos
(alcohol, acetona, éter, cloroformo,
etc.).
Su importancia biológica estriba en que es imposible
vivir sin su aporte, constituyendo la reserva energética más importante
del organismo (los animales de experimentación que son sometidos a dietas
exclusivas en proteínas e hidratos de carbono fallecen cuando han consumido su
tejido adiposo). Además de su principal función energética, con un valor
calórico elevado (9cal/gramo), las grasas también poseen otras misiones
esenciales como: impedir las pérdidas de calor, proteger las vísceras (epiplon,
grasa perivisceral, etc.), transportar vitaminas liposolubles (A, D, E y K) y
ácidos grasos esenciales (linoleico, linolénico y araquidónico).
Entre sus propiedades está el dar a los preparados
culinarios unas características organolépticas especiales, que aumentan su
sabor.
La población del mundo industrializado
occidental, con alta capacidad adquisitiva, ingiere a menudo una cantidad
excesiva, sobre todo de las grasas de origen animal, lo que puede ocasionar
obesidad y enfermedades relacionadas con la arteriosclerosis.
1.
DIGESTION
Y ABSORCION DE LIPIDOS
Previamente
a la digestión enzimática debe producirse la emulsión de las grasas a partir de
las sales biliares, las cuales facilitan la digestión.
Los
triacilgliceridos son el mayor componente energético en la dieta humana y de
los animales superiores en general. Sin embargo, estos lípidos no pueden
atravesar libremente las membranas celulares, lo que se agrava en las células
epiteliales del intestino, los enterocitos, por la presencia de una capa de
agua inmóvil que rodea las microvellosidades. Para que se produzca la correcta
asimilación de los lípidos, estos deben ser hidrolizados por distintas enzimas
digestivas intestinales hasta formar componentes antipáticos, que si podrán atravesar
las membranas celulares, principalmente al nivel del yeyuno. Pero previamente a
la digestión enzimática, debe producirse la emulsión de las grasas por acción
de las sales biliares, las cuales facilitan la digestión. Debido a la actividad
detergente de las sales biliares, las grandes gotas lipídicas de la dieta se
transforman en numerosas gotitas de pequeño tamaño (micelas), consiguiendo
aumentar enormemente la superficie; esto facilita la actuación de las enzimas
digestivas.
Los
triacilgliceridos son digeridos por la lipasa pancreática hasta formar
compuestos anfipáticos, que pueden atravesar las membranas del enterocito.
Estas enzimas hidrolizan los triacilgliceridos de la dieta dando, por cada
molecular inicial, un monoacilglicerol y dos moléculas de ácidos grasos, aunque
pueden liberar glicerol en algunos casos. Dichas sustancias ya son anfipáticas
y pueden atravesar con facilidad las membranas celulares, pudiendo ser
asimiladas por las células de la mucosa intestinal. Una vez dentro de la célula,
los lípidos son reconstruidos en triacilgliceridos.
Sobre
los fosfolípidos actúa la fosfolipasa A2, liberando un ácido graso y un acil
lisofosfolipido; mientras que, sobre los esteres de colesterol, interviene la
colesterol esterasa rindiendo colesterol y ácidos grasos. Todos estos
compuestos anfipáticos son asimilados por los enterocitos y, de igual manera
que se ha descrito con los triacilgliceridos, en su interior, se regeneran los
lípidos iniciales. Para poder ser transportados al resto del organismo, los
lípidos no pueden estar en forma libre, sino que deben constituir un complejo
estable uniéndose a las apoproteinas para originar las llamadas lipoproteínas.
En el intestino se origina, principalmente, un tipo de lipoproteína denominada
quilomicrón.
2.
LIPOPROTEÍNAS
Las
lipoproteínas viajan por la linfa y la sangre
Las
lipoproteínas son unas estructuras complejas que sirven para transportar los
lípidos por el organismo, a nivel sanguíneo y linfático. También colaboran en
el transporte de aminoácidos. La disposición típica de una lipoproteína esta
formada por una capa externa constituida por fosfolípidos, apoproteinas y
colesterol libre, de naturaleza anfipática, mientras que en el interior se
acumulan los triacilglicéridos y el colesterol esterificado, compuestos
totalmente hidrofóbicos. La principal lipoproteína del intestino es el
quilimicrón. Los quilomicrones son vertidos a la linfa y, vía linfática, son
transportados hasta la sangre, de tal forma que llegan primero a los tejidos
periféricos y posteriormente al hígado. Este orden facilita que las grasas se
almacenen en los tejidos periféricos, preferentemente en el músculo y en el
tejido adiposo.
Tipos y función de las
lipoproteínas.
Existen
distintos tipos de lipoproteínas: quilomicrones (QM) , VLDL (lipoproteínas de
muy baja intensidad), IDL (lipoproteínas de densidad intermedia), LDL
(lipoproteínas de baja densidad), HDL (lipoproteínas de alta densidad).
Se
diferencian principalmente por su densidad y tamaño, de modo que su nombre
deriva de esta propiedad. Las diferencias de densidad permiten su aislamiento
fácilmente mediante técnicas de ultracentrifugación o de electroforesis (fig.
14-3). En la tabla 14-1 se presentan
los principales parámetros de las lipoproteínas, tanto físicos como químicos y,
además, los de los constituyentes de cada una de ellas. Como se observan
en la tabla, a la vez que el porcentaje
de triacilglicéridos disminuye, va aumentando el porcentaje de proteínas y de
colesterol libre o esterificado. Este hecho es uno de los factores que
determinan que la densidad de las lipoproteínas vaya en aumento.
En
el intestino se forman gran cantidad de quilomicrones, pero también se pueden
originar pequeñas cantidades de VLDL. Estas lipoproteínasse vierten a la linfa,
que las transporta hasta la sangre, sin pasar por la circulación enterhepática,
de tal forma que llegan primero a los tejidos periféricos (figura 14-4). En estos tejidos la enzima lipoproteína lipasa plasmática ataca a los triglicéridos,
hidrolizándolos en glicerol y ácidos grasos, que son asimilados por las células
tisulares, principalmente adipocitos y miocitos, gracias a que reconocen a la
apo C-11, típica de los quilomicrones y las VLDL. El glicerol y los ácidos
grasos por difusión simple a las células de los tejidos periféricos y son
utilizados para formar triacilglicéridos y almacenar asi grandes cantidades de
energía cuando sea necesaria. De esta forma se produce el transporte de los
triacilglicéridos de la dieta (TG-exógenos) hasta los tejidos. Los restos de
los quilomicrones que quedan tras la actuación
de la lipoproteína lapasa plasmática se conocen como quilomicrones remanentes, pobres en triacilglicéridos pero no en
fosfolípidos y apoproteínas; estos restos son retirados por el hígado,
suministrándose así fosfolípidos, colesterol, ácidos grasos y aminoácidos al
tejido hepático. Con las VLDL ocurre un proceso similar, aunque estas
lipoproteínas suelen ser de origen hepático y transportan los triacilglicéridos
propios del organismo, sintetizados a nivel hepático (TG endógenos).
La
actuación de la lipoproteína lipasa sobre las VLDL origina igualmente glicerol
y ácidos grasos que serán asimilados por los tejidos. Además se generan las IDL
o VLDL remanentes, que son ricas en colesterol; estas lipoproteínas residuales
son retiradas igualmente por el hígado, que las usa como fuente de fosfolípidos,
colesterol y aminoácidos.
A
nivel sanguíneo, también aparecen las HDL, que tienen un origen principalmente
hepático y sirven para recoger el exceso de colesterol depositado en los
tejidos periféricos y transportarlo al hígado. De la misma manera, esta
lipoproteína interviene intercambiando apoproteínas y colesterol esterificado
con las demás lipoproteínas, principalmente las LDL. Hay que destacar que las
HDL tienen un papel importante en la esterificación del colesterol catalizada
por la enzima lecitina colesterol aciltransferasa (LCAT), que esterifica el
colesterol que las HDL tienen un papel importante en la esterificación del
colesterol que las HDL han recogido. (Feduchi, 2010, págs. 257-260)
3.
METABOLISMO
DE ACIDOS GRASOS
LIPOLISIS
La
lipolisis es el mecanismo de movilización de los lípidos que se encuentran
almacenados como reservorio de energía. Esta movilización sucede cuando hay una
deficiencia del aporte energético o cuando se ayuna. Estos lípidos acumulados
en forma de triacilgliceridos se encuentran en forma anhidra, como gotitas de
grasa, en el citoplasma de las células adiposas. El primer paso para su
catabolismo es la hidrolisis por medio de la triacilgliceridos: glicerol y tres
ácidos grasos. La enzima triglicérido lipasa actúa bajo una estrecha regulación
hormonal: el glucagón y la adrenalina potencian sus actividades. Favoreciendo
la lipolisis al fosforilar a la triglicérido
lipasa a través de la proteína quinasa a dependiente de AMPc; mientras que
la insulina, al potenciar una fosfatasa que desfosforila la lipoproteína
lipasa, bloquea la lipolisis.
Los
ácidos grasos salen del adipocito y se unen en sangre a la albumina. Esta
proteína plasmática (que también se conoce como VHDI, lipoproteínas de muy alta
densidad) transporta típicamente entre dos y cuatro moléculas de ácidos grasos,
si bien puede llegar a transportar hasta seis. Estos ácidos grasos movilizados
llegan transportados por la albumina, hasta los tejidos que requieran energía:
típicamente, el hígado, el musculo cardiaco y el musculo esquelético. En estos
tejidos los ácidos grasos serán oxidados en una vía metabólica muy importante,
denominada B-oxidación, para producir grandes cantidades de energía. Otra
fuente importante de ácidos grasos son los fosfolípidos, componentes de la
membrana celular. Estos lípidos estructurales están sometidos a una renovación
continua y, por tanto, su degradación y síntesis son constantes.
El
glicerol también sale a la sangre, pues en el tejido adiposo no puede
metabolizarse; de la sangre es retirado por el hígado, donde se transforma en
dihidroxiacetona fosfato gracias a la actuación secuencial de la glicerol
quinasa, enzima no presente en los adipocitos, y la glicerol-3-P
deshidrogenasa. La dihidroxiacetona fosfato suele entrar en la gluconeogénesis
a nivel hepático, aunque también puede seguir la vía glacolítica sirviendo para
la producción de energía.
4.
DEGRADACION
DE ACIDOS GRASOS
La
B-oxidación
Esta
ruta fue postulada por Knoop en 1904 y confirmada por Leloir, lehninger y
lynen. En la B-oxidación se produce sucesivas oxidaciones en el carbono B, que
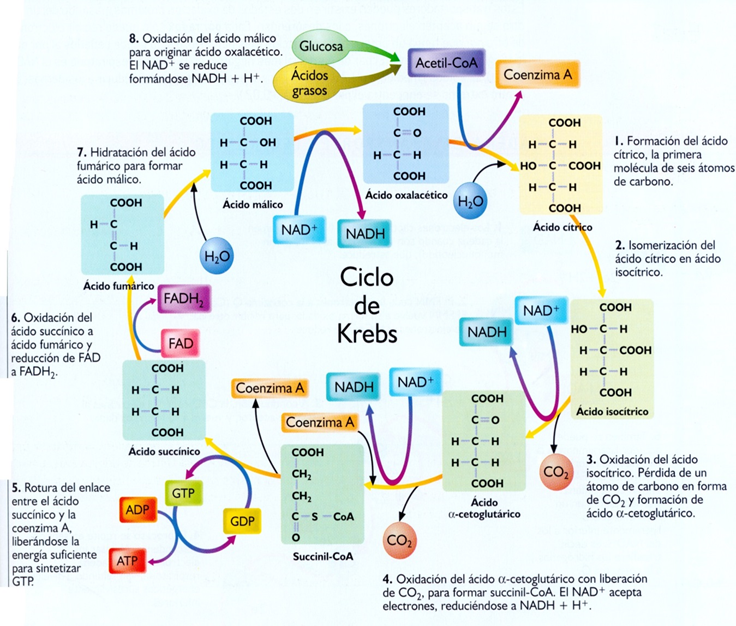
van separado fragmentos de dos carbonos en forma de acetil CoA, que se
incorporaran después al ciclo de Krebs. Al tiempo se producen, tanto en la B-oxidación como en el ciclo de Krebs,
coenzimas reducidas que serán reoxidadas en la “cadena respiratoria” rindiendo
energía en forma de ATP, La B-oxidación tiene lugar en la matriz mitocondrial,
por lo tanto es necesario que el ácido graso penetre en este orgánulo. Así, se
puede dividir la oxidación de los ácidos grasos en tres fases. La primera fase
implica la activación del ácido graso esterificándose con el CoA y a expensas
del ATP: la segunda fase supone la entrada en la mitocondria, gracias a un
transporte mediado por carnitina: y la tercera fase la B-oxidación propiamente
dicha, degradándose el ácido graso a moléculas de acetil CoA.
Los
ácidos grasos que entran en la célula, rápidamente van a ser transformados en
sus correspondientes éster tiolico con la CoA, con la finalidad de activar el
compuesto y solubilizarlo mejor en el entorno celular. Estas transformaciones
esta catalizada por la acil CoA sintetasa: en un primer paso, la enzima produce
la adenilacion del ácido graso formado el acil adenilato, que permanece unido a
la enzima, y el pirofosfato, que se hidroliza; posteriormente, el ácido graso
se transfiere a la molécula de CoA, formando el acil CoA, con la consiguiente
liberación de AMP. Como se puede apreciar, la formación de un éster tiolico
necesita mucha energía; tanta, que implica la hidrolisis de ATP a AMP, además
de la hidrolisis subsiguiente del pirofosfato. La acil CoA sintetasa se
encuentra localizada en la membrana de RE y en la membrana externa de la
mitocondria. La enzima de la membrana del RE activa los ácidos grasos que se
incorporaran en la biosíntesis de lípidos, mientras que la enzima de la
membrana externa de la mitocondria activa los ácidos grasos que entraran en el
interior de la mitocondria para su degradación en la B-oxidación. La actuación
de esta última enzima forma las moléculas de acil CoA en el espacio
intermembrana de la mitocondria.
Sin
embargo, las largas moléculas de acil CoA no pueden entrar en la mitocondria,
al no poder atravesar su membrana interna. Logran penetrar ayudadas por un
sistema de lanzadera: el resto de ácido graso se transfiere a una transportador
denominado carnitina para formar un intermediario acil-carnitina, gracias a la
acción de la carnitina acil transferasa-I. La acil-cartinitina puede atravesar
las membranas mitocondriales debido a la presencia de un transportador
específico: la carnitina acil-cartnitina translocasa, que se localiza en la
membrana interna mitocondrial. Este transportador, a la vez que introduce la
acil-carnitina en el interior mitocondrial, saca carnitina al espacio
intermembrana. Ya en la matriz, el resto de ácido graso es cedido a una molécula CoA, en una reacción
catalizada por la carnitina acil transferasa-II, quedando la carnitina
disponible nuevamente para salir al espacio intermembrana e introducir nuevos
ácidos grasos al interior de la mitocondrial. Este mecanismo de transporte al
interior mitocondrial tiene como finalidad mantener aisladas las moléculas de
CoA de la mitocondria de las del resto de la célula, de tal manera que la relación
existente entre CoA libre y la acil CoA o acetil CoA sirve como indicador del
nivel energético de la mitocondria y permiten un mejor control del gasto
metabólico.
Una
vez dentro de la matriz mitocondrial, las moléculas de acil CoA comienzan
propiamente el proceso degradativo de la B-oxidación. Este proceso se basa en
cuatro pasos que repiten consecutivamente hasta que roda la molécula de acil
CoA ha sido degradada en moléculas de acetil CoA que, finalmente, entraran en
el ciclo de Krebs produciendo más energía. Los cuatro pasos que repiten en el
ciclo son:
1.
DESHIDROGENACION: gracias a la actuación de la enzima acil CoA deshidrogenasa
se introduce un doble enlace trans (entre los carbonos a y B del ácido graso)
obteniéndose una molécula con poder reductor FADH2, y originando una molécula
de enoil CoA.
2.
HIDRATACION: la molécula de enoil CoA se transforma en un hidroxiacil CoA,
mediante la incorporación de una molécula de agua por acción de enoil CoA
hidratasa: el OH del agua en la posición B, y el H en la posición a.
3
DESHIDROGENACION: gracias a la hidroxiacil CoA deshidrogenasa se oxida la
molecula hasta una molécula de cetoacil CoA, oxidando el grupo hidroxilo a un
grupo ceto. Esta oxidación sirve para reducir una molécula de NAD+ A NADH+H+.
4
RUPTURA tiolica: catalizada por una tiolasa y con la intervención de una
molecula de CoA libre. Se genera una molecula de acetil CoA y un acil CoA que
tiene dos carbonos menos en su cadena que el original. El acetil CoA se
incorporara al ciclo de Krebs mientras que el acil CoA acortado en dos carbonos
inicia una nueva “vuelta” en la B-oxidación. El ciclo se repite tantas veces
como sea necesario hasta “cortar” totalmente la cadena de ácido graso en
fragmentos de acetil CoA de dos carbonos.
Al
final, todos los productos originados en la B-oxidación se aprovechan en la
mitocondria para rendir más energía. Por cada vuelta en la B-oxidación, un
ácido graso rinde una molecula de NADH+H+, una molecula de FADH, y una molecula
de acetil CoA. Además en la última vuelta se genera no una, sino dos moléculas
de acetil CoA. Las moléculas de NADH+H y FADH2 entraran en la cadena
transportadora de electrones donde se oxidaran para producir energía en forma
de ATP, mientras que las moléculas de acetil CoA se degradaran en el ciclo de
Krebs originando GTP y más moléculas de poder reductor (NADH+H+ y FADH2) que
también se utilizaran para la síntesis de ATP a través de la fosforilación
oxidativa. Para tener más información sobre la degradación de los ácidos grasos
de cadena impar y los ácidos grasos insaturado.
OXIDACIONES SECUNDARIAS DE LOS ÁCIDOS
GRASOS.
Existen
diversas variantes de la B-oxidación, con la finalidad de cubrir diferentes
necesidades celulares. Por ejemplo en los peroxisomas se origina una variante
en la que la acil CoA deshidrogenasa transfiere los electrones al oxigeno
formando peróxido de hidrogeno empleado en esos orgánulos como agente oxidante
y para facilitar su actividad degradativa. Esta B-oxidación presenta
especificidad por ácidos grasos de cadena larga.
Existen
otras rutas para degradar ácidos como puede ser la a-oxidación y la
o-oxidación. En estas rutas la oxidación va precedida de la hidroxilacion de un
carbono mediante una oxidasa de función mixta (rectículo endoplásmico liso,
mitocondria o peroxisomas). Cuando se produce la hidroxilacion del carbono a
seguida de oxidación a carbonilo y de la descarboxilacion del C-1 en forma de
CO2 se habla de la a-oxidación. Dicha ruta es importante en la oxidación de
ácidos grasos metilados, como el ácido fitanico. El déficit de esta vía produce
la enfermedad de Refsum, trastorno neurológico congénito muy grave, que
origina, entre otras complicaciones, retintis pigmentosa, sordera, ataxia
cerebelosa y neuropatía periférica. Cuando se produce la hidroxilacion del
último carbono, seguida de la oxidacion secuencial a aldehído y a carboxilo, se
habla de oxidacion. Por estas rutas se forman ácidos dicarboxílicos que pueden
entrar en la B-oxidacion por ambos lados, degradándose más rápidamente.
5.
LA
BIOSINTESIS DE ACIDOS GRASOS
Puesto
que la capacidad de los animales para almacenar glucosa es bastante limitada,
la tura biosintetica que conduce desde la glucosa a los ácidos grasos es una
vía muy importante. La glucosa ingerida en exceso se convierte en ácidos grasos
y estos, a su vez, en triacilgliceridos que pueden almacenarse en grandes
cantidades en el tejido adiposo.
Después
del descubrimiento de la B-oxidacion en las mitocondrias y el papel que
desempeña el acetil CoA, se supuso que la biosíntesis de ácidos grasos
consistiría en una simple inversión de las mismas etapas enzimáticas. Sin
embargo, se hicieron algunas observaciones que no estaban de acuerdo con este
punto de vista: un hecho importante es que un citoplasma sin mitocondrias no
están presentes. Además se precisa CO2 que se incorpora al malonil CoA, pero no
se incorpora al ácido graso.
Para
proceder a la síntesis de los ácidos grasos se requiere disponibilidad de poder
reductor, NADHPH+H, que se obtiene de la ruta de las pentosas fosfato y de la
actuación de la enzima málica. Además se necesitan suficientes moléculas de
acetil CoA citoplasmicas, pues la síntesis de los ácidos grasos tienen lugar en
el citoplasma, para lo cual las moléculas de acetil CoA tienen que salir de la
mitocondria, donde se generan a partir del piruvato. Como las moléculas de
acetil CoA no pueden atravesar las membranas de la mitocondria, recurren a un
transporte con citrato y piruvato como vía de salida, transporte conocido como
ciclo del piruvato-citrato. Las moléculas de acetil CoA se utilizan para
sintetizar malonil CoA a través de la enzima acetil CoA carboxilasa, que emplea
como cofactor biotina en un proceso que requiere energía procedente del CO2. La
formación de malonil CoA es el paso clave de regulación, regulación que
principalmente tiene lugar a nivel hormonal.
En
la formación de los nuevos ácidos grasos interviene la ácido graso sintetasa,
complejo multienzimatico que desempeña todas las funciones necesarias para
formar un nuevo ácido graso a partir de moléculas de maloni CoA, NADPH+H y una
molecula de acetil CoA. La ácido graso sintetasa habitualmente va a
sintetizar siempre el mismo ácido graso,
el ácido palmítico, que, posteriormente, se transformara en los demás ácidos
grasos que necesite la célula, mediante enzimas de tipo clongasas y
desaturasas. El ciclo de la ácido graso sintetasa es muy similar a la
B-oxidacion, si bien se realiza en sentido contrario y, en vez de usar la CoA
como transportador de ácidos grasos usa una proteína, la denominada proteína
portadora de grupos acilo (ACP)
El
proceso se inicia con la transferencia del grupo acetilo desde la acetil CoA al
brazo de oscilación de fosfopanteteina de la ACP (ScH: SH periférico).
Posteriormente, al grupo acetilo se une un nuevo resto acetilo, procede de una
molecula de malonil CoA que se había dispuesto en el SH central. La unión
ocurre mediante una condensación en la cual se produce la descarboxilacion del
malonil CoA y deja el residuo de acetil CoA, que se unirá al primer fragmento
de acetil CoA, de modo que se formara un ácido graso de dos átomos de carbono
más, oxidado en el carbono B. a continuación, y de una forma inversa a la
B-oxidacion, el grupo ceto se reduce a un alcohol, se elimina una molecula de
agua formando un doble enlace entre los carbonos a y B, y se satura el doble
enlace mediante otra reducción.
6.
CUERPOS
CETONICOS
Los
cuerpos cetonicos, acetoacetato , hidroxibutirato y acetona , son sustancias
que se producen a partir del acetil CoA en las mitocondrias del tejido hepático
, cuando la velocidad de B –oxidacion supera la velocidad de oxidación del
acetil CoA en el ciclo de krebs , por ejemplo en situaciones de ayuno . Estos
compuesto, que se pueden distribuir a través del sistema circulatorio por todos
los tejidos. Así , favorecen un ahorro de glucosa , glucosa que es fundamental
para otra serie de tejidos que dependen más estrechamente de este hidrato de
carbono para obtener energía como , por ejemplo , el cerebro y los glóbulos
rojos , Incluso si se produce un ayuno muy prolongado puede ser utilizados por
el cerebro como fuente de energía alternativa a la glucosa , Estos compuestos
se utilizan ya que los animales no pueden transformar de forma neta los ácidos
grasos en hidratos de carbono , al carecer del denominado ciclo del glioxilato
El
ciclo del glioxilato es una ruta que se dan en plantas y microorganismos. Estas
ruta se localiza en unos orgánulos subcelulares , conocidos como glioxisomas .
Es una vía alternativa del metabolismo de la acetilo ( en forma de acetil CoA ,
es una molécula de succinato . El succinato es un intermediario del ciclo de
krebs que puede originar fácilmente oxalacetato , moléculas que por
glucogénesis permite formar la glucosa .
Esta ruta permite fijar de forma neta los átomos de carbono de ácidos grasos en
azucares . Los animales al carecer de este ciclo , no pueden realizar dicha
conversación
Esta
vía emplea enzimas del ciclo de kresh , la citrato sintasa y la conitasa ,
además de dos enzimas exclusivas de este ciclo del glioxilato : la isocitrato
liasa y la malato sintasa ( véase figura ). En este ciclo del glioxilato , el
oxalacetato se condensa con el acetil CoA originando citrato . El citrato se
transformara en isocitrato que se escinde en 2 moléculas originando sucionato y
glioxilato , mediante la isocitrato liasa . El glioxilato se condensara con
otro acetil CoA formando el malato por acción de la malato sintasa y finalmente,
el malato regenerara el oxacelato . El succionato formando por la actuación del
isocitrato liasa va a salir del glioxisoma , originando oxacelatato , que puede
transformarse en glucosa vía de la gluconeogenesis . El ciclo de glioxilato
permite la conservación de acetil CoA en glucosa, por lo tanto permite la
conversión de forma neta de los ácidos grasos en glucosa
CETOGENESIS
El
proceso de la creación de los cuerpos ceronicos se conoce como citogénesis.
Básicamente consiste en la condensación de 2 moléculas de acetil CoA por acción
de una tiolasa, formando el acetoacetil CoA . Posteriormente se fusiona una
nueva molécula de acetil CoA, gracias a la acción de la enzima
hidroximetilglutaril CoA sintasa, originando el hidroximetilglutaril CoA,
gracias a la accion de la enzima hidroxmetilglutaril CoA sintasa , originando
el hidroximetilglutaril CoA , Este compuesto sirve para la síntesis de cuerpo
cetonico y también se utiliza para la biosíntesis del colesterol ( véase mas
adelante .) El hidrozimetilglutaril CoA se escinde en acetil CoA se escinde en
acetil CoA y en acetoacetato , el primer cuerpo cetonico , por acción de la
hidroximetiglutaril CoA liasa . El acetoacetato es el precursor de los demás
cuerpos cetonicos que existen en el organismo . Así por reducción , se origina
el Hidroxibutirato , es una reacción catalizada por hidroxibutirato
deshidrogenasa , Por descarboxilacion del acetoacetato se forma acetona ; ; si
bien este paso puede ser realizado enzimaticamente , suele ocurrir de forma
cinéticamente espontánea y de ciertas forma no deseada . La descarboxilacion
implica la perdida de un átomo de carbono , tal manera que la acetona pierde la
capacidad energética y va a rendir menos ATP que el hidroxibutirato y el
acetoacetato . Posteriormente estos compuestos salen de mitocondria atraviesan las células hepática hasta llegar
a la sangre , que se encarga de distribuirlos por todos el organismo.
UTILIZACIÓN DE LOS CUERPOS CETONICOS
Los
cuerpos cetonicos que asimilados por el tejido extrahepáticos se utilizan para
producir molécula de acetil CoA que serán degradados en el ciclo de krebs . El
hidroxibutirato se oxida a acetoacetato , originando NADH+ H que será utilizado
para producir ATP mediante la fosforilación oxidativa en la cadena
transportadora de electrones .El acetoacetato se unirá a CoA formando gracias a
la enzima cetoacetil CoA por un tiolasa rendira 2 moléculas de acetil CoA que
se degradaran posteriormente en el ciclo de los ácidos tricarbocilicos ,
generando gran cantidad de energía . La acetona , debido a que ha perdido un
átomo de carbono de propanodiol y aunque también se puede romper
originando acido formico y acido acético
. Cualquiera de estas 2 posibilidades
aporta menos energía que la aporta la degradación del hidroxibutirato y el
acetoacetato .
El
uso de los cuerpos cetonicos depende de la fluctuaciones de los niveles de
glucosa en sangre . Así después de las comidas cuando la cantidad de glucosa es
elevada todos los tejidos incluidos en músculo liso , usan la glucosa como
principal fuente de energía . En este momento el hígado aprovecha para
almacenar glucosa en forma de glucosa . Cuando los niveles empiezan a decender
, el hígado intenta mantener los niveles de glucosa liberando las reservas que
había almacenado en forma de glucosa , a través de la gluconeogenesis . A su
vez ciertos tejidos como el músculo cardiaco y esquelético , comienza a usar
como principal fuente de energía los ácidos grasos procedentes de la lipólisis
del tejido adiposo , favoreciendo un menor consumo de glucosa . Esto ácidos
grasos también emplea el hígado para realizar la B-oxidación obtener gran cantidad de moléculas de acetil CoA y
con ellas generar los cueros cetonicos . Los cuerpos cetonicos son enviados a
la sangre para que también sirvan de fuente de energía a diversos tejidos
principalmente y el tejido muscular . Cuando el proceso de inanición o ayuno es
muy prolongado y se agotan las reservas de glucogenos decayendo los niveles de
glucosa en sangre de forma importante , la gran mayoria de los tejidos pasa a
alimentarse de ácidos grasos y cuerpo cetonicos . Incluso el cerebro puede
adaptarse y utilizar los cuerpos cetonicos como fuente de energia , en parte
debido a que no puede aprovechar el consumo de glucosa , que queda reservada
casi exclusivamente para los glóbulos rojos m los cuales , al carecer de núcleo
, solo pueden realizar la glucólisis para obtener energía . Además , el
hígado sigue generando pequeñas
cantidades de glucosa a través de la gluconeogenesis principalmente a partir
son generados por una deficiencia absoluta o relativa de insulina ,
amplificados por un incremento en los niveles de las hormonas antiinsulina ,
principalmente glucagon y cortisol.
7.
BIOSÍNTESIS
DE LÍPIDOS
Debido
a la gran heterogeneidad estructural que presentan, la biosíntesis de lípidos
abarca gran cantidad de rutas y procesos metabólicos. Este apartado se centrará
en la síntesis de dos compuestos sumamente importantes: los triacilglicéridos,
que son la forma de almacenamiento a largo plazo preferida por los organismo
superiores (principalmente mamíferos, entre los que se encuentra el hombre) y
el colesterol, compuesto de gran importancia para las membranas celulares
animales y por su pared como precursor de hormonas esteroideas, ácidos biliares
y vitamina D.
LA BIOSÍNTESIS DE LOS ACILGLICÉRIDOS
La
síntesis de los triacilglicéridos, que tiene lugar en el retpiculo endoplásmico
liso (REL) de células adiposas y hepáticas, se origina mediante la
esterificación secuencial de una molécula de glicerol-3-fosfato con tres
moléculas de acil CoA (ácidos grasos activados). Requiere la formación previa
de un fosfolípido intermediario, el ácido fofatídico, compuesto por dos ácidos
grasos, glicerol y un grupo fosfato. El proceso de síntesis del ácido
fosfatídico se puede dividir en diversas etapas:
Síntesis
de glicerol-3-fosfato a partir de glicerol por la acción de la glicerol quinasa
(principalmente en hígado y riñón) o mediante la reducción de la
dihidroxiacetona fosfato a glicerol-3-fosfato por catálisis de la glicerol-3-P
deshidrogenasa, reacción típica de los adipocitos.
Activación
de los ácidos grasos, por defecto de la acil CoA sintetasa, tal y como se
describió en la activación de los ácidos grasos durante la B-oxidación.
Transferencia
de los ácidos grasos activados para originar el ácido fosfatídico, gracias a la
actuación de acil transferasas que transfieren los ácidos grasos de la moléculas
de acil CoA a la posiciones 1 y 2 del glicerol-3-P.
El
ácido fosfatídico originado no sólo sirve para la síntesis de
truacilglicéridos, sino también para la síntesis de fofoglicéridos. En la
formación de un triacilglicérido, el ácido fosfatídico debe desprenderse del
grupo fosfato presente en la posición 3, proceso que ocurre gracias a la acción
de una fosfatasa. La ácido fosfatídico fosfatasa deja, tras su acción un
diacilglicerol. El diacilglicerol se transformará en triacilglicerol mediante
la acil transferasa que transferirá un ácido graso procedente de una molécula
de acil CoA a la posición 3. Finalmente, los triacilgliceroles pueden
almacenarse en el citoplasma en grandes gotas, como ocurre en los adipocitos, o
incorporarse en vesículas de secreción: en lipoproteínas, en intestino e
hígado, o leche en la glándula mamaria.
LA BIOSÍNTESIS DEL COLESTEROL
La
síntesis de colesterol tiene lugar en el citoplasma a partir de moléculas de
acetil CoA, y se puede dividir en tres fases:
Primera
etapa: síntesis de los isoprenos activados a partir de acetil CoA.
Esta
fase comienza con un mecanismo análogo a la síntesis de cuerpos cetónicos, por
la que a partir de tres moléculas de acetil CoA se obtiene una molécula de
hidroximetilglutaril CoA (HMG CoA). Posteriormente el grupo carboxilo del HMG,
que está formado un tiéster con la coenzima A, se reduce a aldehído y después a
un alcohol. El NADPH+H+ es el agente reductor en las dos etapas de la reacción,
originando el mevalonato. La reacción de reducción está catalizada por la HMG
CoA reductasa y es la etapa limitante de la síntesis del colesterol. La enzima
está muy regulada y es una diana farmacológicamente importante.
El
mevalonato es activado hasta dar los isoprenos activados, isopentil-pirofosfato
y dimetilalil-pirofosfato, paso que implica la descarboxilación del grupo ácido
del mevalonato y el gasto de tres moléculas de ATP, dos para la formación del
pirofosfato y una tercera para favorecer la descarboxilación del ácido. Estas
unidades de ispreno activadas son las moléculas claves en la biosíntesis de
gran número de moléculas, entre ellas el colesterol y los terpenos.
Segunda etapa: condensación de seis
moléculas de isoprenos activados para formar escualeno (C30). A partir
de la condensación de una molecula de isopentil-pirofosfato y otra
dimetilalil-pirofosfato se origina el geranil-pirofosfato; el cual se condensa,
a su vez, con otra molécula de isopentil-pirofosfato dando a lugar al
farnesil-pirofosfato. La condensación posterior de dos moléculas de farnesil-pirofosfato,
de tres isoprenos cada una, genera el escualeno.
Tercera etapa: ciclación de escualeno a
lanosterol (C30) y conversión final a colesterol (C22). La
formación del núcleo esteroideo a partir del escualeno comienza con la
formación del epóxido de escualeno. Este intermediario se protona para formar
un carbocatión que se cicla para formar una estructura tetrecíclica que, a su
vez, se reorganiza para formar el lanosterol. El lanosterol se convierte en
colesterol mediante un proceso de múltiples pasos, 19 etapas en total, que
comprende la eliminación de tres grupos metilo, la reducción de un doble enlace
por el NADPH y la migración del otro doble enlace. Hay que destacar que algunas
de las reacciones están catalizadas por enzimas de la superfamilia del
citicromo P450, enzimas que son oxidasas de función mixta y que tienen un papel
muy importante en la destoxificacion de fármacos sobre todo de origen
esteroideo.
En
los vertebrados, la síntesis de colesterol está controlada principalmente
mediante la velocidad a la que el colesterol entra en las células procedente
del torrente sanguíneo. La homeostasis se mantiene mediante un mecanismo que
coordina el consumo de colesterol en el alimento, la síntesis de colesterol
endógeno en el hígado (y, en menor grado, en el intestino)y la tasa de
utilización de colesterol por las células. En este mecanismo interviene el
receptor de LDL, que es la lipoproteína principal encargada del transporte de
colesterol en el torrente sanguíneo.
La
HMG CoA reductasa es la enzima clave de la síntesis de colesterol endógeno y se
controla de múltiples maneras:
·
La velocidad de la síntesis del mRNA de la
reductasa está controlada por la proteína que se une al elemento regulador de
esteroides (SREBP, del inglés Steroid Regulatory Elemente Binding Proteín).
Este factor de transcripción se une a una secuencia corta de DNA, denominada
elemento regulador de esteorides (SER). Los niveles bajos de colesterol
producen la activación de SREBP por degradación proteolítica y migración al
núcleo, donde se une al SER del gen de la HMG CoA redcutasa, así como a otros
genes de la vía biosíntetica del colesterol para aumentar su transcripción.
Cuando aumenta la concentración de colesterol se bloquea la liberación
proteolítica de la SREBP y se degrada la que existe en el núcleo. Estos dos
hechos detienen la transcripción de los genes de la vía biosintética del
colesterol.
·
La velocidad de la traducción del mRNA de
la reductasa se inhibe por metabolitos no esteroles derivados del mevalonato,
así como por el colesterol de la dieta.
·
La degradación de la reductasa está
controlada de modo riguroso, de tal manera que la cantidad de enzima se puede
regular de una a 200 veces. La degradación se estimula por colesterol,
mavalonato, farnesol y derivados del colesterol.
·
La fosforilacion disminuye la actividad de
la reductasa. El proceso se regula a nivel hormonal, de forma que el glucagón
favorece la forma fosfotilada inactiva de la HMG CoA reductasa, mientras que la
insulina induce la forma desfosforilada de la HMG CoA reductosa, que es
inactiva. (Elena, 2010)
Bibliografía
Elena, F. (2010). BIOQUIMICA. PANAMERICANA.
Feduchi, E. (2010). Bioquimica
conceptos esenciales (Primera ed.). Madrid: Panamericana.